Diverse mutazioni nella CMT1X e diversa gravità dei sintomi
Un recente articolo sull’European Journal of Neurology prova a correlare le diverse mutazioni responsabili della CMT1X alla gravità dei sintomi
La malattia di Charcot-Marie-Tooth di tipo X (CMTX1) è una neuropatia ereditaria causata da mutazioni nel gene GJB1, che codifica per la proteina connessina 32, una proteina che regola la permeabilità della membrana cellulare e la comunicazione tra cellule adiacenti. È la seconda forma più comune di CMT e colpisce principalmente i nervi periferici, con sintomi che includono debolezza muscolare e perdita di sensibilità. Gli uomini sono generalmente più gravemente colpiti rispetto alle donne.
In un recente lavoro scientifico, un equipe di medici francese ha recentemente studiato numerosi pazienti con CMT1X per evidenziare eventuali correlazioni tra le varianti genetiche (mutazioni) specifiche del gene GJB1 e le manifestazioni cliniche della malattia, al fine di migliorare la formazione di gruppi di pazienti omogenei per futuri studi clinici.
Risultati Principali
Lo studio ha analizzato 275 pazienti affetti da CMTX1 provenienti da 13 centri di riferimento in Francia, raccogliendo dati genetici, clinici e neurofisiologici. Ecco i risultati principali:
- Varianti Genetiche: Sono state identificate 87 varianti distinte del gene GJB1. La maggior parte di queste varianti erano missense, ma sono state trovate anche varianti nonsense, frameshift e nella regione non codificante 5’UTR.
- Correlazione Fenotipo-Genotipo: I pazienti con varianti nei domini transmembrana della proteina connessina 32 hanno mostrato sintomi più gravi, un esordio della malattia più precoce e una maggiore perdita di funzione motoria rispetto a quelli con varianti nei domini intracellulari o extracellulari.
- Implicazioni Cliniche: La gravità dei sintomi varia significativamente in base al dominio della proteina colpito dalla mutazione. Questo suggerisce che il genotipo potrebbe avere un ruolo prognostico oltre che diagnostico.
Esiste una correlazione tra gravità della malattia e mutazione nella CMT1X?
Lo studio condotto da Luce Barbat du Closel e colleghi conferma l’esistenza di una correlazione significativa tra il dominio proteico mutato e il fenotipo clinico nei pazienti con CMTX1. In particolare, i pazienti con varianti nei domini transmembrana della proteina connessina 32 hanno manifestato sintomi più severi rispetto a quelli con varianti nei domini intracellulari o extracellulari.
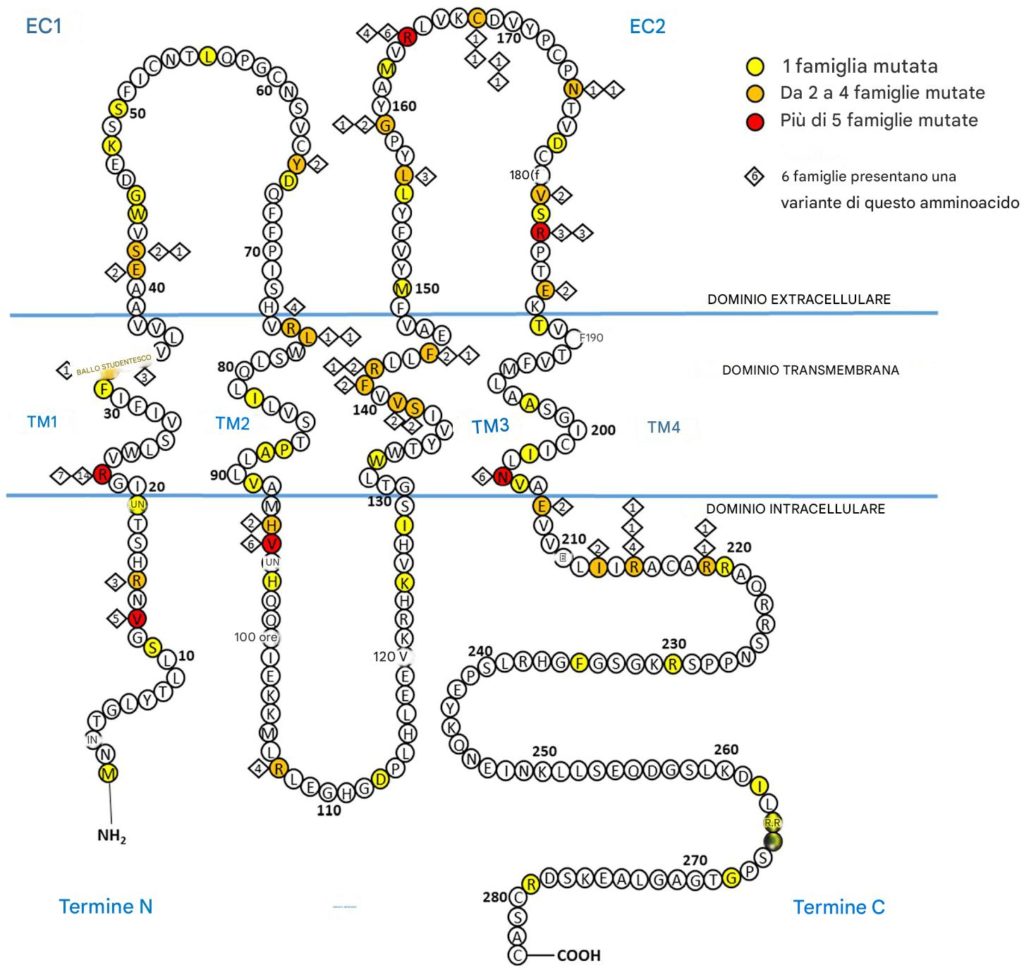
Questo risultato è cruciale per diverse ragioni:
- Prognosi e Diagnosi: La scoperta che il dominio proteico mutato può influenzare la gravità dei sintomi suggerisce che il genotipo potrebbe essere utilizzato non solo per diagnosticare la malattia, ma anche per prevederne l’andamento. Questo potrebbe migliorare significativamente la consulenza genetica fornita ai pazienti e alle loro famiglie.
- Progettazione di Studi Clinici: La comprensione delle correlazioni fenotipo-genotipo è essenziale per la formazione di gruppi di pazienti omogenei nei futuri studi clinici. Questo è particolarmente importante per le sperimentazioni di nuove terapie, dove la variabilità dei sintomi tra i partecipanti può influenzare i risultati. La possibilità di formare gruppi di pazienti con caratteristiche cliniche simili migliorerà l’accuratezza e l’affidabilità dei risultati degli studi.
- Sviluppo di Terapie Personalizzate: La conoscenza delle specifiche varianti genetiche che influenzano la gravità della malattia può guidare lo sviluppo di terapie mirate. Ad esempio, i pazienti con varianti nei domini transmembrana potrebbero beneficiare di trattamenti specifici che tengano conto della maggiore severità dei loro sintomi.
- Inclusione nei Trial Clinici: Lo studio suggerisce che i pazienti con varianti di significato incerto (VUS) o con mutazioni nella regione non codificante 5’UTR non mostrano differenze cliniche significative rispetto ad altri gruppi. Pertanto, potrebbe essere prudente includere questi pazienti nei futuri trial clinici, ampliando così il pool di partecipanti eleggibili e migliorando la rappresentatività dei risultati.
In sintesi, questo studio rappresenta un passo avanti significativo nella comprensione della malattia di Charcot-Marie-Tooth di tipo X e offre importanti spunti per la diagnosi, la prognosi e il trattamento di questa malattia.
Fonte: “Phenotype–genotype correlation in X-linked Charcot-Marie-Tooth disease: A French cohort study”, pubblicato sull’European Journal of Neurology nel 2024.